
Flu Vaccines Contain RNA That Trigger Positive PCR Test Results: ‘Journal of Medical Microbiology’
Is the “chilling” rise in flu cases nationwide attributable to PCR tests detecting vaccine RNA, not wild virus?

Mainstream news outlets are broadcasting that there is a “chilling” rise in flu cases, with Colorado, Louisiana, and New York experiencing the “fastest increases in influenza cases.”
However, the rise in cases follows flu vaccination campaigns in those states, which raises questions about vaccine efficacy.
But it also raises questions about whether the vaccinations themselves are contributing to the increasing case numbers.
For example, the New Orleans Health Department (NOHD) launched a flu vaccination campaign in early October.
NYC Health Department similarly launched an October push urging all residents 6 months and older to get flu shots.
The Colorado Department of Public Health and Environment’s (CDPHE) influenza webpage was updated the same month to promote flu vaccination.
These campaigns are meant to increase flu vaccine uptake.
Now there’s a rise in influenza cases, which are counted using positive PCR test results.
However, a March 2012 Journal of Medical Microbiology publication confirms the presence of residual viral RNA (genomic RNA—which PCR tests look for—from the influenza viruses used in vaccine production) in inactivated split-virus seasonal influenza vaccines.
One of the most popular injectable flu vaccines in the U.S., the formaldehyde-containing ‘Fluzone High-Dose,’ is an inactivated split-virus vaccine.
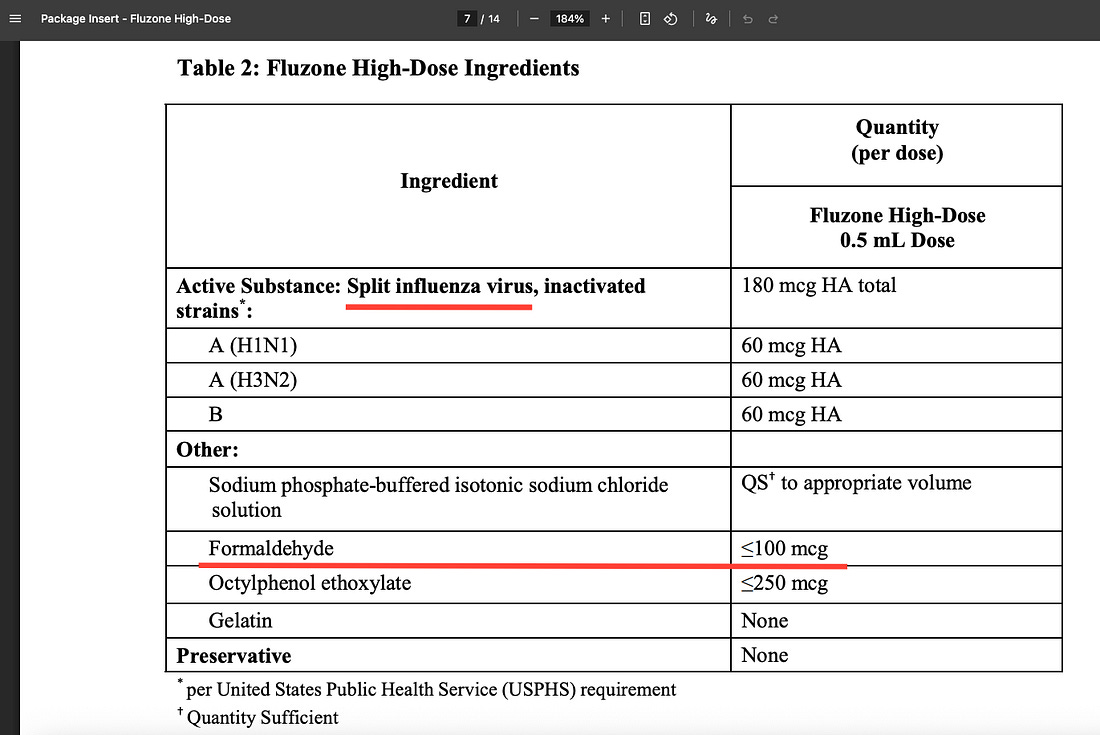
The 2012 study directly tested two 2010 trivalent inactivated vaccines (egg-based, similar in type to Fluzone) and detected high quantities of influenza A and B viral RNA using real-time RT-PCR on the vaccine liquid itself.
This RNA was stable, remaining detectable for at least 66 days after opening the vials.
Sequencing confirmed it included genetic components matching vaccine strains.
The study abstract reads:
False-positive PCR results usually occur as a consequence of specimen-to-specimen or amplicon-to-specimen contamination within the laboratory. Evidence of contamination at time of specimen collection linked to influenza vaccine administration in the same location as influenza sampling is described. Clinical, circumstantial and laboratory evidence was gathered for each of five cases of influenza-like illness (ILI) with unusual patterns of PCR reactivity for seasonal H1N1, H3N2, H1N1 (2009) and influenza B viruses. Two 2010 trivalent influenza vaccines and environmental swabs of a hospital influenza vaccination room were also tested for influenza RNA. Sequencing of influenza A matrix (M) gene amplicons from the five cases and vaccines was undertaken. Four 2009 general practitioner (GP) specimens were seasonal H1N1, H3N2 and influenza B PCR positive. One 2010 GP specimen was H1N1 (2009), H3N2 and influenza B positive. PCR of 2010 trivalent vaccines showed high loads of detectable influenza A and B RNA. Sequencing of the five specimens and vaccines showed greatest homology with the M gene sequence of Influenza A/Puerto Rico/8/1934 H1N1 virus (used in generation of influenza vaccine strains). Environmental swabs had detectable influenza A and B RNA. RNA detection studies demonstrated vaccine RNA still detectable for at least 66 days. Administration of influenza vaccines and clinical sampling in the same room resulted in the contamination with vaccine strains of surveillance swabs collected from patients with ILI. Vaccine contamination should therefore be considered, particularly where multiple influenza virus RNA PCR positive signals (e.g. H1N1, H3N2 and influenza B) are detected in the same specimen.
The human body’s own extracellular vesicles (EVs) (including exosomes) naturally carry and transfer various RNAs (here, here, here, here) through bodily fluids such as blood, saliva, urine, nasal mucus, and cerebrospinal fluid.
These are the exact substances PCR tests are applied to.
Another popular flu vaccine is FluMist, whose FDA package insert directly confirms uses a live virus that can be shed from bodily fluids for at least 28 days after vaccination, detectable with PCR tests.
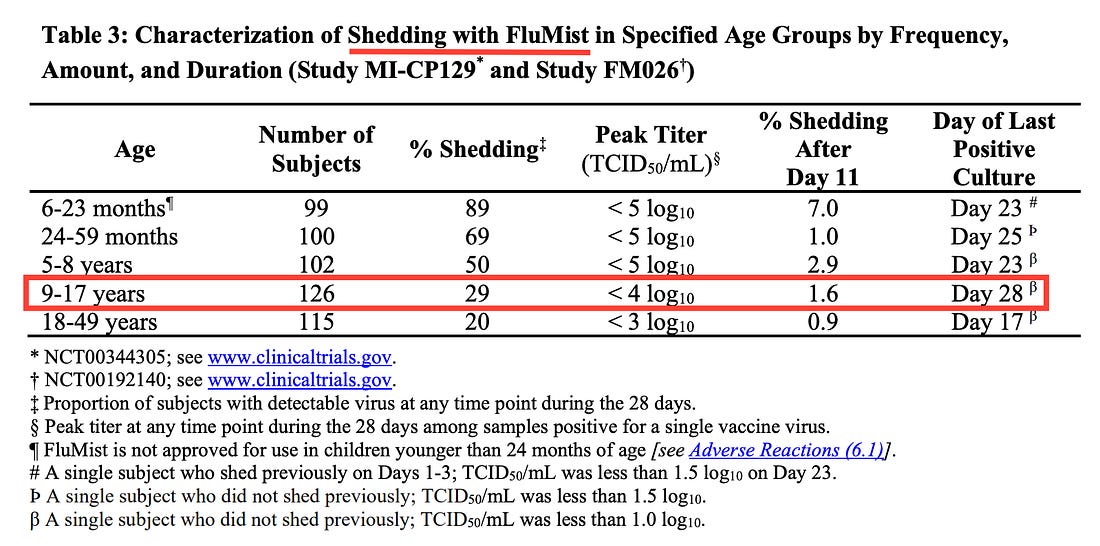
All together, this data raises logical questions:
- Are PCR tests detecting vaccine virus RNA, not wild virus RNA?
- Is the nationwide rise in flu-positive PCR tests attributable, at least in part, to the detection of vaccine material?
- Why haven’t the CDC or vaccine manufacturers directly tested this?


Recent Comments